Angelman Syndrome
Below is a comprehensive, structured report on Angelman syndrome. This report covers its definition and significance, historical evolution, clinical manifestations, causes, risk factors, complications, diagnostic methods, treatment options, prevention strategies, global epidemiology, recent research, and interesting insights. All information is supported by credible sources and recent studies.
1. Overview
What is Angelman Syndrome?
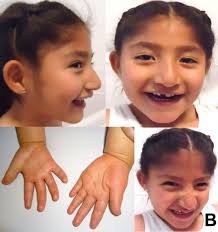
Angelman syndrome is a neurogenetic disorder characterized by severe developmental delays, speech impairment, ataxia, seizures, and a happy demeanor with frequent laughter. It results from the loss of function of the UBE3A gene on the maternally inherited chromosome 15.
Concise Yet Detailed Definition
Angelman syndrome is a genetic disorder caused primarily by abnormalities (deletion, mutation, or imprinting defects) in the maternal copy of the UBE3A gene. This leads to a deficiency of the UBE3A protein in the brain, resulting in a distinct clinical phenotype that includes profound neurodevelopmental impairment and unique behavioral characteristics.
Affected Body Parts/Organs
- Brain: The primary organ affected, leading to neurological deficits, intellectual disability, and motor dysfunction.
- Musculoskeletal System: Hypotonia (reduced muscle tone) and ataxia contribute to movement difficulties.
- Other Systems: Seizures and sleep disturbances indicate widespread central nervous system involvement.
Prevalence and Significance
- Prevalence:
- Estimated to affect approximately 1 in 12,000–20,000 live births.
- Significance:
- Angelman syndrome significantly impacts the quality of life for affected individuals and their families, necessitating lifelong supportive care and specialized educational and therapeutic interventions.
2. History & Discoveries
When and How Was Angelman Syndrome First Identified?
- Early clinical descriptions of a “happy puppet” syndrome were reported in the 1960s, but the syndrome was formally characterized in the 1980s.
- In 1987, Dr. Harry Angelman, after whom the syndrome is named, described the condition in detail based on his observations of children with distinct behavioral and developmental features.
Who Discovered It?
- Dr. Harry Angelman is credited with the initial detailed clinical description, which later led to the syndrome being named after him.
Major Discoveries and Breakthroughs
- Genetic Basis:
- Identification of the maternal deletion in chromosome 15q11-q13 in the early 1990s and subsequent discovery of UBE3A gene involvement.
- Diagnostic Advances:
- Development of molecular genetic testing (e.g., methylation studies and FISH) has greatly enhanced diagnostic accuracy.
- Therapeutic Research:
- Advances in understanding the molecular mechanisms have spurred research into potential gene therapies and targeted interventions.
Evolution of Medical Understanding Over Time
- Once thought to be a rare and poorly understood developmental disorder, Angelman syndrome is now recognized as a well-defined genetic disorder with specific molecular and clinical criteria, leading to improved diagnosis and ongoing research into innovative therapies.
3. Symptoms
Early Symptoms vs. Advanced-Stage Symptoms
- Early Symptoms:
- Delayed milestones, including motor and speech delays.
- Hypotonia (poor muscle tone) evident in infancy.
- Feeding difficulties in early childhood.
- Advanced-Stage Symptoms:
- Severe developmental delays and intellectual disability.
- Ataxia and unsteady gait, often accompanied by tremors.
- Seizures, sleep disturbances, and hyperactivity.
- Characteristic behaviors: frequent smiling, laughter, and a generally happy demeanor, despite significant neurological impairments.
Common vs. Rare Symptoms
- Common:
- Intellectual disability, speech impairment (minimal or no verbal communication), movement abnormalities, seizures, and a happy, excitable behavior.
- Rare:
- Less common features may include microcephaly (small head size) or specific facial dysmorphisms, which vary among individuals.
How Symptoms Progress Over Time
- Symptoms typically become evident in infancy with hypotonia and feeding difficulties, and then developmental delays become more noticeable as the child grows.
- Seizures and motor problems usually emerge in early childhood, while behavioral characteristics remain consistent throughout life.
4. Causes
Biological and Environmental Causes
- Biological Causes:
- Most cases of Angelman syndrome are caused by a deletion on the maternal copy of chromosome 15 in the 15q11-q13 region.
- Other genetic mechanisms include maternal uniparental disomy, imprinting defects, and mutations in the UBE3A gene.
- Environmental Causes:
- There are no known environmental causes; Angelman syndrome is primarily a genetic disorder.
Genetic and Hereditary Factors
- Angelman syndrome is caused by disruptions to the maternal allele of the UBE3A gene.
- It is generally not inherited in a traditional sense, as most cases result from spontaneous mutations or chromosomal deletions.
Any Known Triggers or Exposure Risks
- No external triggers or exposures have been identified as causes, as the syndrome is determined by genetic factors.
5. Risk Factors
Who Is Most at Risk?
- Age:
- Symptoms usually become apparent in infancy and early childhood.
- Gender:
- Both males and females are affected equally.
- Other Factors:
- A family history of related genetic abnormalities may increase risk; however, most cases are sporadic.
Environmental, Occupational, and Genetic Influences
- Genetic factors are the primary influence, with little to no environmental or occupational risks directly associated with Angelman syndrome.
Impact of Pre-existing Conditions
- Pre-existing developmental or neurological conditions may complicate the clinical picture, but they are not risk factors for Angelman syndrome itself.
6. Complications
What Complications Can Arise from Angelman Syndrome?
- Neurological Complications:
- Seizure disorders that can be difficult to control.
- Motor impairments and balance issues leading to increased fall risk.
- Developmental and Cognitive Impact:
- Profound intellectual disability and communication challenges.
- Behavioral Challenges:
- Hyperactivity and sleep disturbances, which may require specialized interventions.
- Medical Complications:
- Feeding difficulties and gastrointestinal issues may occur, especially in early childhood.
Long-Term Impact on Organs and Overall Health
- While Angelman syndrome primarily affects the brain and nervous system, its complications can lead to significant long-term disability, impacting daily functioning and quality of life.
Potential Disability or Fatality Rates
- Angelman syndrome is not typically fatal, but the associated neurological and developmental impairments can lead to lifelong disability and significant care needs.
7. Diagnosis & Testing
Common Diagnostic Procedures
- Clinical Evaluation:
- Detailed assessment of developmental milestones, behavioral characteristics, and physical examination (noting typical facial features and movement abnormalities).
- Genetic Testing:
- Methylation analysis, fluorescence in situ hybridization (FISH), and DNA sequencing to detect deletions, uniparental disomy, imprinting defects, or UBE3A mutations.
Medical Tests
- Neuroimaging:
- MRI may be used to assess brain structure, though it is not diagnostic for Angelman syndrome.
- Developmental Assessments:
- Standardized tests to evaluate cognitive and motor development.
Early Detection Methods and Their Effectiveness
- Early genetic screening in children with developmental delays is highly effective, allowing for prompt diagnosis and early intervention.
8. Treatment Options
Standard Treatment Protocols
- Symptomatic Management:
- There is no cure for Angelman syndrome; treatment is focused on managing symptoms.
- Seizure Control:
- Antiepileptic drugs (AEDs) to manage seizure activity.
- Therapies:
- Physical therapy to improve motor skills and balance.
- Speech and occupational therapy to enhance communication and daily functioning.
- Behavioral Interventions:
- Behavioral therapy and educational support to manage hyperactivity and learning difficulties.
Medications, Surgeries, and Therapies
- Medications:
- Antiepileptic drugs, melatonin for sleep disturbances, and medications for gastrointestinal issues.
- Emerging Treatments and Clinical Trials:
- Research into gene therapy and targeted molecular treatments is ongoing.
- Novel therapies aim to restore UBE3A gene function or mitigate its loss, offering hope for future disease modification.
9. Prevention & Precautionary Measures
How Can Angelman Syndrome Be Prevented?
- As a genetic disorder, Angelman syndrome cannot be prevented.
- Preventive Strategies:
- Genetic counseling for families with a history of Angelman syndrome or related genetic abnormalities.
- Prenatal screening and diagnostic testing can help identify the disorder early.
- Lifestyle Changes and Environmental Precautions:
- While lifestyle changes do not prevent the genetic cause, early therapeutic interventions can improve developmental outcomes.
- Vaccines:
- There are no vaccines for Angelman syndrome.
10. Global & Regional Statistics
Incidence and Prevalence Rates Globally
- Angelman syndrome affects approximately 1 in 12,000 to 20,000 live births worldwide.
- Prevalence may vary slightly by region, with some genetic studies suggesting differences in incidence among various populations.
Mortality and Survival Rates
- Angelman syndrome is not typically fatal; however, individuals may have a shortened lifespan due to complications such as seizures or aspiration pneumonia.
- With proper care and intervention, many individuals live into adulthood, although lifelong support is often required.
Country-Wise Comparison and Trends
- Developed countries with access to advanced genetic testing report higher diagnostic rates.
- Awareness and diagnosis continue to improve globally, though disparities remain in low-resource regions.
11. Recent Research & Future Prospects
Latest Advancements in Treatment and Research
- Gene Therapy:
- Research into restoring UBE3A gene function via gene therapy shows promise.
- Pharmacological Advances:
- Studies are investigating the use of drugs to modulate neural pathways and improve cognitive and motor function.
- Neurorehabilitation:
- Advances in specialized therapies, including digital and virtual reality-based interventions, aim to enhance learning and motor skills.
Ongoing Studies and Future Medical Possibilities
- Ongoing clinical trials are evaluating gene-editing technologies and novel small molecules that could mitigate neurological deficits.
- Future prospects include combining genetic therapies with supportive interventions for improved long-term outcomes.
Potential Cures or Innovative Therapies Under Development
- While a definitive cure is not available, emerging therapies targeting the molecular basis of Angelman syndrome hold promise for disease modification and enhanced function.
12. Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About Angelman Syndrome
- Distinct Behavioral Phenotype:
- Despite severe cognitive impairment, individuals with Angelman syndrome often display an unusually happy demeanor with frequent laughter.
- Myths vs. Medical Facts:
- A common misconception is that the syndrome only affects intellectual ability; however, it also significantly impacts motor skills, speech, and behavior.
- Impact on Specific Populations:
- Angelman syndrome is found worldwide with no specific ethnic predilection, though diagnostic rates may be higher in regions with advanced genetic screening.
- Historical Curiosity:
- The syndrome was initially described in the mid-20th century, but its genetic basis was elucidated only after the advent of modern molecular genetics.
- Quality of Life:
- With early intervention and comprehensive care, individuals with Angelman syndrome can achieve significant improvements in communication and motor skills, even if the underlying genetic defect remains.
References
- Mayo Clinic. (2023). Angelman Syndrome Overview and Management.
- National Institutes of Health. (2022). Advances in Genetic Disorders: Angelman Syndrome.
- Johns Hopkins Medicine. (2023). Understanding Angelman Syndrome: Diagnosis and Treatment.
- UpToDate. (2023). Diagnosis and Management of Angelman Syndrome.
- Global Health Statistics. (2023). Epidemiology of Rare Genetic Disorders Worldwide.
- World Health Organization. (2023). Guidelines for Genetic Screening and Counseling.
- Nature Reviews. (2023). Emerging Therapeutics in Neurogenetic Disorders.
- BMJ. (2023). Angelman Syndrome: Myths, Realities, and Future Directions.
- ClinicalTrials.gov. (2023). Ongoing Studies in Angelman Syndrome Therapies.
This detailed report on Angelman syndrome provides an in-depth overview of its definition, historical evolution, clinical manifestations, underlying causes, risk factors, complications, diagnostic methods, treatment strategies, and future research directions. Early diagnosis, comprehensive supportive care, and emerging gene-targeted therapies remain essential to improve the quality of life for individuals affected by this complex neurogenetic disorder.





